
Los recursos computacionales de Argonne respaldaron el análisis completo más grande de COVID-19 secuencias del genoma en los EE. UU. y ayudó a corroborar la creciente evidencia de una mutación proteica.
antes de COVID-19-19 ingresó por primera vez a los Estados Unidos en marzo, el Hospital Metodista de Houston ya había comenzado los preparativos para probar y secuenciar el virus a gran escala, dadas las noticias provenientes de Wuhan, China.
Entre marzo 5, cuando apareció el primer caso en el área metropolitana de Houston, y julio 7, médico / investigadores de Houston Methodist secuenciaron el genoma de más 5,085 cepas del virus. Estos representaron casi 10 por ciento de la COVID-19-19 casos que pasaron por el 2,400en el sistema de salud Metodista de Houston, durante dos oleadas distintas que ocurrieron en ese período de tiempo.
“El 99 por ciento no es el 100 por ciento. Si hay una mutación que representa solo el uno por ciento de la población y suprime o erradica a la mayoría, puede aumentar algún rasgo de ese uno por ciento, ya sea virulencia o transmisibilidad, y entonces es un juego de pelota diferente “. – James Davis, científico del personal de Argonne
Colaboradores de la Universidad de Texas en Austin, Weill Cornell Medical College, la Universidad de Chicago y el Departamento de Energía de EE. UU. (GAMA) El Laboratorio Nacional Argonne trabajó en conjunto para analizar los datos y tratar de correlacionar los resultados del paciente con los rasgos virales.
“Este es el análisis de secuencia viral más grande en los EE. UU. En este momento y es una de las instantáneas continuas más completas de secuencias que data del comienzo del brote ”, dijo James Davis, científico de planta de la división de ciencia de datos y aprendizaje de Argonne.“También proporciona una imagen mucho más clara de cómo están evolucionando las cepas “.
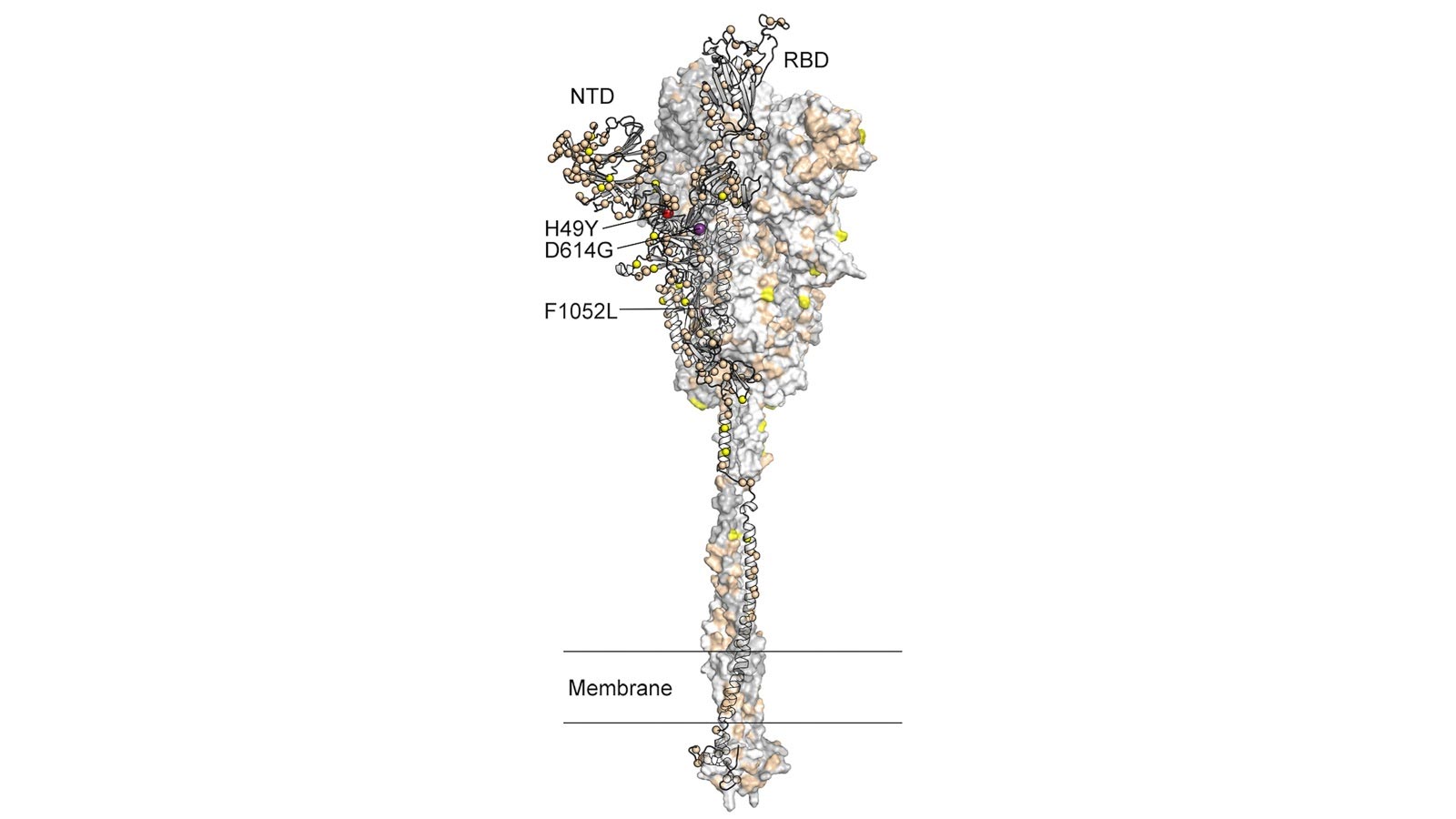
Durante el curso de la investigación, el grupo ayudó a solidificar las crecientes observaciones y preocupaciones a nivel internacional de que una mutación en la proteína del pico del virus se había vuelto dominante, lo que COVID-19-19tasas de transmisibilidad como lo atestiguó la segunda ola que atravesó Houston a mediados de mayo.
Se publicó en la revista un artículo que describe sus métodos y resultados mBio en octubre 30, 2020.
Esa mutación en el pico, responsable de infiltrarse en el sistema inmunológico humano y el objetivo actual de la investigación de vacunas, estaba en un amino ácido llamado Gly614 y fue el resultado de una proteína, el ácido aspártico, que se transformó en otra, la glicina.
Durante las primeras partes de la pandemia, de marzo a abril, Gly614 era solo una variante entre muchas otras. Pero durante la segunda ola en mayo, recuerda Davis, todos los casos que estaban secuenciando en Houston Methodist mostraron que Gly614 había proliferado hasta el punto de convertirse en el aminoácido dominante en la proteína de pico.
De hecho, se encontró en más 99 porcentaje de las variantes secuenciadas.
“El SARS-CoV-2 El virus se conserva notablemente, por lo que cada vez que ve cambios como este es más notable porque no tiende a ver tantas mutaciones ”, dijo.“No estoy seguro de si hace que el virus sea más virulento o más fácil de transmitir, pero el estudio muestra algunos datos que sugieren que los pacientes con Gly614 mutaciones tienen una carga viral mayor, aunque no necesariamente están más enfermas “.
Coincidente con el Gly614 En la segunda ola, los pacientes tendían a ser más jóvenes, mostraban síntomas menos graves, tenían más probabilidades de ser hispanos / latinos y vivían en áreas de ingresos medios más bajos. Aún así, las razones no estaban claras y esperaban que los recursos computacionales de Argonne abrieran una puerta a las causas.
Una relación de trabajo ya establecida, Houston Methodist se acercó a Argonne para obtener ayuda con los análisis de la secuencia del genoma del 5,000-más COVID-19 cepas, así como análisis filogenéticos, que analizan los cambios en un organismo o una característica específica a lo largo del tiempo.
A través de su Proyecto del Centro de Recursos Bioinformáticos, respaldado por el Instituto Nacional de Alergias y Enfermedades Infecciosas, Argonne proporciona recursos informáticos y técnicos a los colaboradores que llevan a cabo grandes proyectos de datos biológicos. En este caso, administró los componentes de secuenciación e incluyó el control de calidad, las alineaciones del genoma y la construcción de árboles filogenéticos.
“Para ser un virus, tiene un genoma bastante grande ”, señaló Davis,“por lo tanto, el proceso se volvió computacionalmente costoso “. Pero con su arsenal de computadoras, tanto grandes como súper, Argonne estaba preparado para manejar la afluencia de datos.
Otro aspecto de ese trabajo involucró la técnica de inteligencia artificial llamada aprendizaje automático. Si bien el enfoque del aprendizaje automático entre muchas instituciones de investigación, incluida Argonne, se ha centrado en determinar cómo las drogas pueden interactuar con COVID-19-19, Marcus Nguyen esperaba predecir si la secuencia del virus podría, en última instancia, predecir el resultado del paciente o la demografía del paciente.
Nguyen, un especialista en investigación con una cita conjunta en Argonne y la Universidad de Chicago, analizó las correlaciones entre las secuencias del genoma y la información del paciente.
El proceso entrenó un algoritmo de aprendizaje automático en las secuencias genómicas de Houston Methodist, así como en los resultados de pacientes pasados (duración de la estadía en el hospital, necesidad de ventilación mecánica, mortalidad) para determinar potencialmente esos resultados.
“Desafortunadamente, no obtuvimos los resultados que esperábamos ”, dijo Nguyen.“Aunque se han encontrado algunas correlaciones en diferentes piezas de metadatos del paciente, no creo que haya nada en el genoma que indique el resultado del paciente, por lo que tiene que haber algo más en juego “.
Si bien el grupo observó otras variaciones en el pico, la investigación continúa sobre Gly614 para comprender su dinámica y determinar qué papel, si lo hay, podría desempeñar en las terapias de tratamiento.
“Debería ser útil en el desarrollo de vacunas porque muestra tantas secuencias diferentes y tantas variantes que se obtiene una imagen bastante clara de qué buscar en términos de las variantes dominantes en la población ”, dijo Davis.
Aunque el virus se ha mantenido algo estable, hasta la fecha, este estudio, y la historia natural, han demostrado que solo se necesita una mutación para crear un impacto poderoso en la vida, tanto grande como diminuta.
“99 el porcentaje no es 100 por ciento ”, señaló Davis.“Si hay una mutación que representa solo el uno por ciento de la población y suprime o erradica a la mayoría, puede aumentar algún rasgo de ese uno por ciento, ya sea virulencia o transmisibilidad, y entonces es un juego de pelota diferente “.
Esta investigación aparece en el artículo“Arquitectura molecular de diseminación temprana y segunda ola masiva del SARS-CoV-2 Virus in a Major Metropolitan Area ”, en mBio, octubre 30, 2020. Maulik Shukla, tanto de Argonne como de la Universidad de Chicago, fue uno de los coautores.
Lea La mutación genética del coronavirus puede haber hecho que COVID-19 sea más contagioso para obtener más información sobre esta investigación.
Referencia: “Arquitectura Molecular de Difusión Temprana y Segunda Ola Masiva del SARS-CoV-2 Virus in a Major Metropolitan Area ”por S. Wesley Long, Randall J. Olsen, Paul A. Christensen, David W. Bernard, James J. Davis, Maulik Shukla, Marcus Nguyen, Matthew Ojeda Saavedra, Prasanti Yerramilli, Layne Pruitt, Sishir Subedi, Hung-Che Kuo, Heather Hendrickson, Ghazaleh Eskandari, Hoang AT Nguyen, J. Hunter Long, Muthiah Kumaraswami, Jule Goike, Daniel Boutz, Jimmy Gollihar, Jason S. McLellan, Chia-Wei Chou, Kamyab Javanmardi, Ilya J. Finkelstein y James M. Musser, 30 de octubre de 2020, mBio.
DOI: 10.1128 / mBio.02707-20
La financiación de la investigación fue proporcionada por los Institutos Nacionales de Salud (NIH) Instituto Nacional de Alergias y Enfermedades Infecciosas (NIAID) Centro de recursos de bioinformática bacteriana y viral.

